Ionic crystals. Kvant
What is ionic polarization
Ionic polarization consists in the displacement of ions in an external electric field and the deformation of the electron shells in this case. Consider a crystal of the $M^+X^-$ type. The crystal lattice of such a crystal can be considered as two cubic lattices, one of which is built from $M^+$ ions, the other from - $X^-$ and they are inserted one into the other. Let us direct an external uniform electric field ($\overrightarrow(E)$) along the Z axis. The ionic lattices will shift in opposite directions on segments $\pm z$. If we assume that $m_(\pm )(\omega )^2_0$ is a quasi-elastic force that returns an ion with mass $m_(\pm )$ to the equilibrium position, then the force ( $F_(upr)$), which is equal to:
In this case, the electric force ($F_e$), which acts on ions of the same lattice, is equal to:
Equilibrium conditions
In this case, the equilibrium conditions will take the form:
For positive ions:
For negative ions:
In this case, the total relative displacement of ions is equal to:
Ionic polarization is:
where $V_0$ is the volume of one molecule.
If we take, for example, the $NaCl$ structure, in which each ion is surrounded by six ions of the opposite sign, which are located at a distance a from it, then we get:
and, therefore, using (5) and (6), we get that:
Ionic polarization is established in a very short time approximately $(10)^(-13)sec.$ It does not lead to energy dissipation, does not cause dielectric losses. When the external field is removed, the electron shells return to their previous state.
The ionic lattice polarization is described by formula (9). In most cases, such polarization is anisotropic.
where $\left\langle \overrightarrow(p)\right\rangle $ is the average value of the dipole moments of ions, which are equal in absolute value but differently directed, $\overrightarrow(p_i)$ are the dipole moments of individual ions. In isotropic dielectrics, the average dipole moments coincide in direction with the strength of the external electric field.
Local field strength for crystals
The local field strength ($\overrightarrow(E")\ or\ sometimes\ \overrightarrow(E_(lok))\ $) for cubic crystals can be expressed by the formulas:
where $\overrightarrow(E)$ is the average macroscopic field in the dielectric. Or:
If equation (10) is applicable for cubic crystals to calculate the local field, then the Clausius-Mossotti formula can be applied to such crystals:
where $\beta $ is the polarizability of the molecule, $n$ is the concentration of molecules.
The relationship between the polarizability ($\beta $) of a molecule and the dielectric susceptibility ($\varkappa$) for cubic crystals can be given by the expression:
Example 1
Task: The dielectric constant of the crystal is equal to $\varepsilon =2.8$. How many times is the local strength ($\overrightarrow(E")$) of the cubic system field greater than the strength of the average macroscopic field in the dielectric ($E$)?
We take as a basis the formula for calculating the local field strength, namely:
\[\overrightarrow(E")=\frac(\varepsilon +2)(3)\overrightarrow(E)\left(1.1\right).\]
Therefore, for the desired ratio of intensities, we can write that:
\[\frac(E")(E)=\frac(\frac(\varepsilon +2)(3)E)(E)=\frac(\varepsilon +2)(3)\left(1.2\right) .\]
Let's do the calculations:
\[\frac(E")(E)=\frac(2,8+2)(3)=1,6.\]
Answer: 1.6 times.
Example 2
Task: Determine the polarizability of carbon atoms in diamond ($\beta $) if the permittivity of diamond is $\varepsilon =5.6$ and its density is $(\rho )_m=3.5\cdot (10)^3\ frac(kg)(m^3.)$
As a basis for solving the problem, we take the Clausius-Mossotti equation:
\[\frac(\varepsilon -1)(\varepsilon +2)=\frac(n\beta )(3)\left(2.1\right).\]
where the particle concentration $n$ can be expressed as:
where $(\rho )_m$ is the mass density of the substance, $\mu =14\cdot (10)^(-3)\frac(kg)(mol)$ is the molar mass of carbon, $N_A=6.02\cdot (10)^(23)mol^(-1)$ is Avogadro's constant.
Then expression (2.1) will take the form:
\[\frac(\varepsilon -1)(\varepsilon +2)=\frac(\beta )(3)\frac((\rho )_mN_A)(\mu )\ \left(2.3\right).\]
From expression (2.3) we express the polarizability $\beta $, we get:
\[\ \beta =\frac(3\mu (\varepsilon -1))((\rho )_mN_A(\varepsilon +2))\left(2.4\right).\]
Substitute the available numerical values, carry out the calculations:
\[\beta =\frac(3\cdot 14\cdot (10)^(-3)(5.6-1))(3.5\cdot (10)^3\cdot 6.02\cdot (10 )^(23)(5,6+2))=\frac(193,2\cdot (10)^(-3))(160,132\cdot (10)^(26))=1,2\cdot ( 10)^(-29)m^3\]
Answer: $\beta =1,2\cdot (10)^(-29)m^3$.
Stasenko A., Brook Yu. Ionic crystals, Young's modulus and planetary masses // Kvant. - 2004. - No. 6. - S. 9-13.
By special agreement with the editorial board and the editors of the journal "Kvant"
The Little Prince lived and lived. He lived on a planet that was slightly larger than himself...
The little prince described everything to me in detail, and I drew this planet.
Antoine de Saint-Exupery. Little Prince
What atoms are planets made of?
Have you ever thought about how different planets differ from each other? Of course, masses and sizes, you say. This is correct, the masses and radii of the planets largely determine their other characteristics. Well, what atoms of chemical elements are the planets built of? Astrophysicists claim that from different. But in the solar system, and in general in the universe, atoms of different elements are far from being present in equal quantities. It is known, for example, that the relative content (by weight) of hydrogen, helium and all other elements is determined by the ratios of 0.73:0.25:0.02.
The planets in our solar system are also built differently. The largest of them are Jupiter and Saturn (their masses, respectively, are 318 and 95 times the mass of the Earth). M h) - mainly consist of hydrogen and helium. True, both hydrogen and helium in these planets are not in a gaseous state, but in a solid or liquid state, and the average densities of these planets far exceed the density of planetary atmospheres or, for example, gases with which we usually experiment when studying gas laws in a physical workshop. . The planets Uranus and Neptune have masses, respectively, 15 and 17 times greater than that of the Earth, and they consist mainly of ice, solid methane ( CH 4 ) and ammonia ( NH3 ) in the metallic phase. Note that when the mass of the planets decreases (if you “move” along the mass scale from the giant planets), the average mass numbers of the atoms from which these planets are built increase. Is it by chance? It seems that not - the same statement turns out to be true with further "movement" along the mass scale. The terrestrial planets (Mercury, Venus, Mars) do not exceed the mass of the Earth, and iron is a characteristic element for them (and for the Earth). In addition, they contain a lot of silicates (for example, silicon dioxide SiO2 ). The trend is quite clear - the greater the mass of the planet, the smaller the average mass numbers of the atoms of which it consists. A rather natural question arises - is it possible to say that there is some connection between the masses of the planets and the masses of the atoms from which they are built?
Of course, it would be wrong to say that the masses of atomic nuclei depend on the mass of the planet. The atoms of each chemical element are arranged in exactly the same way, not only on different planets, but in general in any place in the Universe. But the connection between the masses of those atoms from which the planets are actually “built” and the masses of the planets themselves really exists. And that's what we're going to talk about next.
We will be discussing a very simple model. But “very often, a simplified model sheds more light on how the nature of a phenomenon actually works than any number of calculations. ab initio for various specific cases, which, even if they are correct, often contain so many details that they obscure rather than clarify the truth. These words belong to the Nobel Prize winner in physics, one of the greatest theoretical physicists of our time, F. Anderson.
Surprisingly, the planets of our solar system, as it turns out, are not so far from the model discussed below. Nevertheless, we must already here warn readers against too formal application of those simple formulas that we write out below, to real planets. All estimates that we will make are valid only in order of magnitude. We will use qualitative considerations and the method of dimensions for estimates, and will not care about those numerical coefficients that arise in more accurate calculations. This approach is justified if the numerical coefficients in the formulas turn out to be of the order of unity. But just such a situation arises in physics and astrophysics quite often (although, of course, not always). There are more serious reasons for this, but we will not discuss them here, but simply accept without proof that dimensionless coefficients will not spoil (at least qualitatively) our conclusions.
On the way to our main goal - establishing a connection between the masses of the planets and their chemical composition - we will make a short excursion into solid state physics and calculate the energy of an ionic crystal and its Young's modulus. Ultimately, these calculations will help us deal with the planets.
Ionic crystals and Young's modulus
Consider first the model of an ionic crystal similar to a salt crystal NaCl , but differing from the latter in that the atoms have approximately the same masses. This is different from the crystal NaCl not very important for further reasoning, but will somewhat facilitate our calculations. We can neglect the mass of electrons in comparison with the mass of atomic nuclei.
Let the crystal density ρ , and the mass numbers of the atoms that make up it, A 1 ≈ A 2 ≈ BUT. The masses of nucleons - protons and neutrons, which make up the nuclei, differ very slightly, we will not take into account the differences between them here. Under these assumptions, we can assume that the mass of each atom is approximately equal to the mass of the atomic nucleus
\(~m \approx Am_p,\)
where m p is the mass of the nucleon. If a unit volume contains only n atoms, then their total mass is equal to the density:
\(~nm = \rho.\)
It is convenient for us to rewrite this simple formula in another way. For the estimates we are about to make, we can consider our model crystal to be cubic. This means that the atoms "sit" in the corners of an elementary cube - a cell of the crystal lattice. Denote the length of the edge of this cube by the letter but. By its very nature, the value n directly related to but\[~na^3 = 1\], so
\(~\rho = \frac(m)(a^3).\)
This formula is curious in that its right-hand side includes m And a- “microscopic” values, on the left is a completely “macroscopic” value - the density of the crystal.
Our crystal lattice is built of alternating positive and negative ions. For simplicity, the charge of each ion will be considered equal to the charge of an electron with the corresponding sign, i.e. ± e. The forces acting on each ion are the usual Coulomb forces. If we had only two ions and they were at a distance a from each other, then the potential energy of their interaction would be \(~\sim \frac(e^2)(\varepsilon_0 a)\), where ε 0 is the electrical constant, and the "~" sign means that we wrote the estimate in order of magnitude. The interaction energy of ion pairs is a very important and useful characteristic for evaluations. But particles in a crystal are, of course, much larger than two. If we assume that the average distance between particles is 2·10 -10 m, then it is easy to calculate that in 1 cm 3 there will be about 10 23 particles.
One often speaks of the density of the electrostatic energy of a system of ions that form a crystal. The word "density" is used here because it refers to the energy per unit volume. In other words, this value is the sum of the potential energies of interaction of all pairs of ions in a unit volume. But it is difficult to accurately calculate such a sum, we will not be able to do this here, because for this it would be necessary to take into account the interaction of a large number of particles located at different distances from each other. It is possible, however, to proceed by analogy with the formula for the density of a crystal.
Note first that the energy density of interest to us w has the dimension J/m 3 , and the dimension of the potential energy of a pair of ions is \(~\left[ \frac(e^2)(\varepsilon_0 a) \right]\) = J. The symbol [...]- denotes the dimension of the quantity in brackets. Let us now divide the "microscopic" quantity \(~\frac(e^2)(\varepsilon_0 a)\) into another, also "microscopic" one - a 3 , and we will get a quantity that has the dimension of energy density. One might think that this is precisely the assessment for w.
These considerations, of course, are not a rigorous proof that the electrostatic energy density of the system of ions forming a crystal is \(~\frac(e^2)(\varepsilon_0 a^4)\). However, an exact calculation for an ionic crystal leads to the formula
\(~w = \alpha n \frac(e^2)(\varepsilon_0 a) = \alpha \frac(e^2)(\varepsilon_0 a^4),\)
which differs from our estimate only by a numerical factor α ~ 1.
The elastic properties of matter are determined, of course, by interatomic interactions. The most important characteristic of such properties is, as we know, Young's modulus E. We are accustomed to defining it from Hooke's law as such a stress at which the relative linear deformation of the body \(~\frac(\Delta l)(l)\) is equal to unity, or, in other words, the corresponding length is doubled. But the value of E does not at all depend on whether we know Hooke's law and whether it actually holds. Let's pay attention to the dimension of the modulus of elasticity: N / m 2 \u003d J / m 3. One can therefore interpret E and as some characteristic energy density.
To make this clearer, let's look at two other examples. The first refers to a conventional flat capacitor. If charges are placed on its plates ± q, then an electrostatic field will exist inside the capacitor, and the plates themselves will be attracted to each other. Let the area of each plate S, and the distance between them d. You can calculate the force of attraction of the plates and, by dividing it by S, find the "characteristic pressure". Or you can calculate the energy contained in the capacitor, and by dividing it by the volume SD, find the energy density. In both cases, the value \(~\frac(\sigma^2)(2 \varepsilon_0)\) is obtained, where \(~\sigma = \frac qS\) is the surface charge density on the plates. The "characteristic pressure" and the energy density in this case turn out to coincide not only in dimensions, but also numerically.
The second example is the determination of the surface tension coefficient of a liquid. This coefficient can be defined as a force per unit length (for example, for a stretched soap film), or it can be considered as the surface energy density. And in this case, the same value is defined in the "power" and "energy" languages.
Let us return, however, to the ionic crystal. The energy characteristic of an ionic crystal is electrostatic energy, the elastic properties of a crystal are determined by the electrical interactions of its constituent particles. Therefore, it can be considered that w ~ E. Here we again assume without proof that the coefficient of proportionality for these quantities is of the order of unity. Thus we have learned evaluate value of Young's modulus for an ionic crystal:
\(~E \sim w \sim \frac(e^2)(\varepsilon_0 a^4) \approx \frac(\rho)(m) \frac(e^2)(\varepsilon_0 \left(\frac( m)(\rho) \right)^(\frac 13)) = e^2 m^(-\frac 43) \rho^(\frac 43) \varepsilon_0^(-1).\)
It immediately follows from this formula that w- value limited from above. As long as it exists ionic lattice, the distance between the ions in any case cannot be less than the size of the atoms (ions). If this were not the case, the electron shells of neighboring ions would overlap, the electrons would become socialized, and instead of an ionic crystal, we would have a metal.
On the other hand, for an ionic crystal, the value w limited from below. You can understand this with this example. Imagine that a deforming force is applied to a crystalline rod. With a sufficiently large value of this force, the rod will collapse. The stress that occurs when breaking is equal to the "breaking" force divided by the cross-sectional area of the rod perpendicular to this force. This tension, let's call it p pr , is called the tensile strength, and it is always less than Young's modulus. The last statement is at least plausible. As we have already said, a stress equal to Young's modulus formally leads to a twofold change in the length of the sample under study. (True, it would be necessary to say also that, generally speaking, it is impossible to use Hooke's law for sufficiently large deformations, but the qualitative conclusions of interest to us still remain without Hooke's law.) We know from experience that to stretch or compress any the crystal is almost impossible to double - it will break long before that. Let now R- characteristic pressure due to external influence on the crystal. We can say that one of the conditions for the existence of a crystal structure is the fulfillment of the inequalities
\(~w > p_(pr) > p.\)
Another obvious condition is the requirement that the temperature of the crystal be less than the melting point of the crystal lattice.
Here another question arises. If Young's modulus is defined as a stress that doubles the length of a rod, then what about a crystal that has the shape of a ball or a cube and is deformed simultaneously from all sides? In this case, it is more reasonable to speak of a relative change not of some length, but volume crystal \(~\frac(\Delta V)(V)\), and Hooke's law for small deformations can be written as
\(~\frac pK = \frac(\Delta V)(V).\)
This formula is very similar to the one we write for the case of tension (or compression) of the rod\[~\frac pE = \frac(\Delta l)(l)\], but Young's modulus E is now replaced by an all-round compression module TO. Module TO can also be interpreted as a characteristic energy density.
Ionic crystal planet
We now turn to our main task. Let's consider a hypothetical planet built of almost identical atoms forming a crystal lattice. For the planet to be entirely crystalline, in any case, it is necessary that the pressure in the center of the planet (it is, of course, maximum there!) Does not exceed the value w.
Pressure at the center of a planet with mass M and radius R can be estimated by the formula
\(~p \sim G \frac(M^2)(R^4),\)
where G is the gravitational constant. This formula can be obtained from dimensional considerations. Recall how this is done.
Suppose that the pressure at the center of the planet can depend on the mass of the planet M, its radius R and gravitational constant G, and write the formula
\(~p \sim G^xM^yR^z.\)
Numbers X, at, z not yet known. Let us write out the dimensions of the parameters included in this formula: [ R] = kg m -1 s -2 , [ G] \u003d m 3 kg -1 s -2, [ M] = kg, [ R] = m. Comparing the dimensions of the left and right parts of the formula, we obtain
Kg m -1 s -2 \u003d m 3x kg -x s -2x kg y m z.
In order for the equality to be fair, it is necessary that the numbers X, at, z satisfied the following system of equations:
\(~\left\(\begin(matrix) 1 = -x + y, \\ -1 = 3x + z, \\ -2 = -2x. \end(matrix) \right.\)
From here X = 1, at = 2, z= -4 and we get our formula for pressure.
On the other hand, this formula can be understood in the following way. Gravitational energy of a ball with mass M and radius R should be of the order \(~\frac(GM^2)(R)\), but we will get the density of gravitational energy if we divide the energy by the volume of the ball V ~ R 3 . Just as elastic moduli can be interpreted as the density of electrostatic energy, the density of gravitational energy can be considered to be of the same order of magnitude as the pressure at the center of a gravitating ball.
We emphasize once again that we are not talking about the identity of pressure and energy density (that would be just a wrong statement!), But about their equality in order of magnitude.
The condition for the existence of an ionic crystal in the center of our hypothetical planet is as follows:
\(~G\frac(M^2)(R^4)< w \sim e^2 m^{-\frac 43} \rho^{\frac 43} \varepsilon_0^{-1}.\)
And, of course, a completely crystalline planet only exists if it is relatively cold, in other words - the temperature at the center of the planet should not be very close to the melting point. Otherwise, the planet would have a liquid core - the crystal would have melted. Consider again that \(~\rho \sim \frac(M)(R^3)\) and \(~m \approx Am_p\), then our inequality can be rewritten as follows:
\(~A< \left(\frac{e^2}{\varepsilon_0 G m_p M} \right)^{\frac 43} \left(\frac{M}{m_p} \right)^{\frac 14}.\)
From this it is already clearly seen that the assumptions that the planet entirely crystalline, and its density in the center is of the order of the average density, lead us to restrictions on the masses of atoms, of which such planets can be built.
The assumption that the average density of the planet coincides in order of magnitude with the density at its center is completely natural and quite reasonable in those cases when the matter in the center of the planet is compressed "not too strongly." But if the compression were very great, the ionic crystal would still no longer exist. If an ionic-crystal planet has the same radius and mass as the Earth, then the density of matter in the center and near the surface differ not so much - only three times. Therefore, in order of magnitude, the average density is indeed the same as the density near the center of the planet. The same is true for not very accurate estimates for other planets and stars.
Limitations on the maximum masses of atoms from which entirely crystalline planets can be built are thus determined by the parameters of the planets themselves. For the simplest model of a continuous ionic-crystal planet, we have obtained
\(~A_(max) = \operatorname(const) \cdot M^(-\frac 12).\)
Let's now draw the graph of the function M(A max) (see figure). This graph, strictly speaking, refers only to our hypothetical situation, when the planets are built of ionic crystals and do not have any significant liquid cores. Recall the beginning of the article, where it was discussed which elements or compounds are characteristic of real planets. Suppose that the planets of the "Solar System" (quotes distinguish hypothetical planets from real ones with approximately the same masses!) Are ionic-crystalline. If we accept that the value of the average mass number for the "terrestrial planets" is about 60, for "Uranus" and "Neptune" about 16, and for "Jupiter" and "Saturn" 2-4, then the corresponding "points" quite well "lay down ' to our schedule. On the horizontal axis on it, we plotted the average value of λ for the "planets", and on the vertical axis - the masses of ionic-crystal planets in units of the mass of the Earth.
a) Dependence of the relative mass of a hypothetical planet on the mass number of atoms; b) the same, but on a logarithmic scale
But this, of course, does not mean that real planets do not have liquid cores - such cores probably exist. However, there are also crystalline structures in the planets. And the fact that real planets, at least qualitatively, are similar to model planets allows us to assert that we really “caught” and understood the regularity of the existence of a connection between the masses of planets and the masses of atoms of the main part of the planet’s constituent substance.
Let us add in conclusion that arguments similar to those given in this article can also be carried out for those cases where the planets are not ionic-crystalline, but metallic. Metallicity means that in a crystal (or in a liquid) there are ions and "free" electrons, torn off at high pressure from "their" atoms. In this case, it is said that the gravitational contraction is "opposed" by the pressure of the electron gas, the balance of the corresponding forces (pressures) ensures the possibility of the existence of stable planets. The principle of calculation leading to the establishment of a connection between the masses of the planets and the characteristics of their constituent atoms remains the same, while the calculations themselves become more complicated, and we will not give them here. For those who wish to do such calculations on their own, we will inform you that the pressure of the electron gas in metals is, in order of magnitude, \(~\frac(\hbar^2)(m_e) n_e^(\frac 53)\), where \(~ \hbar\) ≈ 10 -34 J s - Planck's constant, m e \u003d 10 -30 kg is the electron mass, and n e is the number of electrons per unit volume.
An ideal ionic crystal consists of positively and negatively charged spherical ions. If not all, then at least some of the alkali halide compounds, i.e., are most consistent with this idea. salts formed by one of the alkali metals (lithium, sodium, potassium, rubidium, cesium) and one of the halogens (fluorine, chlorine, bromine, iodine). There is evidence that the crystals of these salts are indeed formed by positive metal ions and negatively charged halogen ions. The most direct of them is the data of X-ray diffraction analysis, on the basis of which the electronic charge distribution is calculated (cm. rice. 9 for the case of NaCl).(22.74 Kb)The fact that such solids are composed of ions rather than atoms can be explained as follows. First of all, all alkali metal atoms have one outer valence electron, while the outer shell of halogen atoms contains seven valence electrons. When a valence electron passes from an alkali metal atom to a halogen atom, two ions are formed, each of which has a stable electronic configuration characteristic of inert gas atoms. Even more important is the energy gain due to the Coulomb attraction between positive and negative ions. Consider sodium chloride (NaCl) as an example. To tear off the outer (valence) electron from the Na atom, you need to spend 5.14 eV (ionization energy). When this electron is attached to the Cl atom, there is an energy gain of 3.61 eV (electron affinity energy). Thus, the energy required for the transition of a valence electron from Na to Cl is (
5,14 - 3.61) eV = 1.53 eV. The Coulomb energy of attraction between two emerging Na ions+ and Cl- at a distance between them (in a crystal) equal to 2.18, is 5.1 eV. This value more than compensates for the total energy of the electron transition and leads to a decrease in the total energy of the system of ions in comparison with a similar system of free atoms. This is the main reason that alkali halide compounds are composed of ions, not atoms.Calculating the energy of ionic crystals is actually more complicated than it might seem from the above discussion. But at least for alkali halide crystals, there is good agreement between the theoretical and experimental values of the binding energy. The ionic bonds are quite strong, as indicated, for example, by the high melting point of 1074 K for NaCl.
Due to the high degree of stability of the electronic structure, ionic crystals fall into the category of dielectrics. Because positive and negative ions interact with electromagnetic waves, ionic crystals exhibit strong optical absorption in the infrared region of the spectrum. (The frequency of the oscillating external electric field in this region of the spectrum is close to the natural frequency of transverse lattice waves, in which the positive and negative ions of the crystal move in opposite directions.) In the visible region of the spectrum, the oscillation frequencies are too high for massive ions to have time to respond to the action of such waves. Therefore, light waves pass through the crystal without interaction, i.e. such crystals are transparent. At even higher frequencies - in the ultraviolet region of the spectrum - field quanta can have sufficient energy to excite valence electrons, which ensures the transition of valence electrons of negative ions to the unoccupied states of positive ions. This leads to strong optical absorption.
covalent crystals. The best known covalent crystals are diamond, silicon and germanium. Each atom in such crystals is surrounded by four neighboring atoms located at the vertices of a regular tetrahedron. Free atoms of each of these elements have four valence electrons, and this is enough to form four paired electronic bonds (between this atom and its four nearest neighbors). Thus, two electrons are collectivized by two atoms that form a bond and are located in space along the line connecting the atoms. This is almost the same bond as between two hydrogen atoms in the hydrogen molecule H 2 . In diamond, these bonds are very strong, and since they have a strictly defined direction relative to each other, diamond is an extremely hard material. The strength of the covalent bond of an electron with a crystal is characterized by the so-called energy gap - the minimum energy that must be transferred to an electron so that it can move freely in the crystal and create an electric current. For diamond, silicon, and germanium, the width of this gap is 5.4, 1.17, and 0.744 eV, respectively. Therefore, diamond is a good dielectric; the energy of thermal vibrations in it at room temperature is too small to release valence electrons. In silicon, and especially in germanium, due to the relatively small width of the energy gap, thermal excitation of a certain number of valence electrons at room temperature is possible. Thus, they conduct current, but since their conductivity is much less than that of metals, silicon and germanium are classified as semiconductors.The ions that make up ionic crystals are held together by electrostatic forces. Therefore, the structure of the crystal lattice of ionic crystals must ensure their electrical neutrality.
On fig. 3.24-3.27 schematically depict the most important types of crystal lattices of ionic crystals and provide detailed information about them. Each type of ion in the ionic lattice has its own coordination number. So, in the crystal lattice of cesium chloride (Fig. 3.24), each Cs + ion is surrounded by eight Cl ions "and, therefore, has a coordination number of 8. Similarly, each Cl- ion is surrounded by eight Cs + ions, i.e. also has a coordination number of 8. Therefore it is assumed that the crystal lattice of cesium chloride has a coordination of 8: 8. The crystal lattice of sodium chloride has a coordination of 6: 6 (Fig. 3.25).Note that in each case, the electrical neutrality of the crystal is maintained.
The coordination and type of crystal structure of ionic lattices are determined mainly by the following two factors: the ratio of the number of cations to the number of anions and the ratio of the radii of cations and anions.
G early centered cubic or octahedral

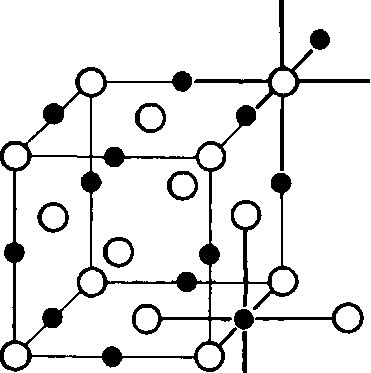
Rice. 3.25. Crystal structure of sodium chloride (rock salt).
The ratio of the number of cations to the number of anions in the crystal lattices of cesium chloride (CsCl), sodium chloride (NaCl) and zinc blende (zinc sulfide ZnS) is 1:1. Therefore, they are classified as stoichiometric type AB. Fluorite (calcium fluoride CaF2) is of the AB2 stoichiometric type. A detailed discussion of stoichiometry is given in Chap. 4.
The ratio of the ionic radius of the cation (A) to the ionic radius of the anion (B) is called the ratio of ionic radii rJrB. In general, the greater the ratio of ionic radii, the greater the coordination number of the lattice (Table 3.8).
Table 3.8. Dependence of coordination on the ratio of ionic radii
Coordination Ratio of ionic radii



Rice. 3.26. Crystal structure of zinc blende.
As a rule, it is easier to consider the structure of ionic crystals as if they consist of two parts - anionic and cationic. For example, the structure of cesium chloride can be thought of as consisting of a cubic cationic structure and a cubic anionic structure. Together they form two interpenetrating (nested) structures that form a single body-centered cubic structure (Fig. 3.24). A structure like sodium chloride, or rock salt, also consists of two cubic structures, one cationic and the other anionic. Together they form two nested cubic structures, forming a single face-centered cubic structure. The cations and anions in this structure have an octahedral environment with a 6:6 coordination (Fig. 3.25).
Zinc blende-type structure has a face-centered cubic lattice(Fig. 3.26). You can think of it as if the cations form a cubic structure, and the anions have a tetrahedral structure inside the cube. But if we consider anions as a cubic structure, then cations have a tetrahedral arrangement in it.
The fluorite structure (Fig. 3.27) differs from those discussed above in that it has the AB2 stoichiometric type, as well as two different coordination numbers - 8 and 4. Each Ca2 + ion is surrounded by eight F- ions, and each F- ion is surrounded by four Ca2 + ions . The structure of fluorite can be thought of as a face-centered cubic cationic lattice, inside which there is a tetrahedral arrangement of anions. It can also be represented in another way: as a body-centered cubic lattice, in which the cations are located in the center of the cubic cell.

Face-centered cubic and body-centered cubic


All compounds considered in this section are assumed to be purely ionic. Ions in them are considered as hard spheres with strictly defined radii. However, as noted in sect. 2.1, many compounds are partly ionic and partly covalent. As a result, ionic compounds with a pronounced covalent character cannot fully obey the general rules set forth in this section.
Such substances are formed with the help of a chemical bond, which is based on the electrostatic interaction between ions. Ionic bond (according to the type of polarity - heteropolar) is mostly limited to binary systems like NaCl(fig.1.10, but), that is, it is established between the atoms of the elements that have the highest electron affinity, on the one hand, and the atoms of the elements that have the lowest ionization potential, on the other. When an ionic crystal is formed, the nearest neighbors of a given ion are ions of the opposite sign. With the most favorable ratio of the sizes of positive and negative ions, they touch each other, and an extremely high packing density is achieved. A slight change in the interionic distance in the direction of its decrease from the equilibrium one causes the appearance of repulsive forces of the electron shells.
The degree of ionization of the atoms that form an ionic crystal is often such that the electron shells of the ions correspond to the electron shells characteristic of rare gas atoms. A rough estimate of the binding energy can be made by assuming that most of it is due to the Coulomb (that is, electrostatic) interaction. For example, in a crystal NaCl the distance between the nearest positive and negative ions is approximately 0.28 nm, which gives the value of the potential energy associated with the mutual attraction of a pair of ions, about 5.1 eV. The experimentally determined energy value for NaCl is 7.9 eV per molecule. Thus, both quantities are of the same order, and this allows using this approach for more accurate calculations.
Ionic bonds are undirected and unsaturated. The latter affects the fact that each ion tends to bring the largest number of ions of the opposite sign closer to itself, that is, to form a structure with a high coordination number. Ionic bonding is common among inorganic compounds: metals with halides, sulfides, metal oxides, etc. The binding energy in such crystals is several electron volts per atom, so such crystals have high strength and high melting points.
Let us calculate the energy of the ionic bond. To do this, we recall the components of the potential energy of an ionic crystal:
Coulomb attraction of ions of different sign;
Coulomb repulsion of ions of the same sign;
quantum-mechanical interaction when electron shells overlap;
van der Waals attraction between ions.
The main contribution to the binding energy of ionic crystals is made by the electrostatic energy of attraction and repulsion, the role of the last two contributions is insignificant. Therefore, if we denote the energy of interaction between ions i And j through , then the total energy of the ion, taking into account all its interactions, will be
Let's provide in the form of the sum of the potentials of repulsion and attraction:
where the plus sign is taken in the case of identical charges, and the minus sign is taken in the case of opposite charges. The total energy of the lattice of an ionic crystal, which consists of N molecules (2 N ions), will be
When calculating the total energy, each interacting pair of ions should be taken into account only once. For convenience, we introduce the following parameter , where is the distance between two neighboring (opposite) ions in the crystal. In this way
where Madelung constant α and constant D are defined as follows:
Sums (2.44) and (2.45) must take into account the contribution of the entire lattice. The plus sign corresponds to the attraction of opposite ions, the minus sign to the repulsion of like ions.
We define the constant as follows. In the equilibrium state, the total energy is minimal. Therefore, , and therefore we have
where is the equilibrium distance between neighboring ions.
From (2.46) we get
and the expression for the total energy of the crystal in the equilibrium state takes the form
The quantity represents the so-called Madelung energy. Since the exponent is , the total energy can be almost completely identified with the Coulomb energy. A small value indicates that the repulsive forces are short-range and change sharply with distance.
As an example, let's calculate the Madelung constant for a one-dimensional crystal - an infinite chain of ions of the opposite sign, which alternate (Fig. 2.4).
Choosing any ion, for example, the “–” sign as the initial one, we will have two ions of the “+” sign at a distance r 0 from it, two ions of the "-" sign at a distance of 2 r 0 and so on.
Therefore, we have
Using the series expansion , we obtain the Madelung constant in the case of a one-dimensional crystal
Thus, the expression for the energy per molecule takes the following form
In the case of a three-dimensional crystal, the series converges conditionally, that is, the result depends on the method of summation. It is possible to improve the convergence of the series if groups of ions are selected in the lattice in such a way that the group is electrically neutral, and if necessary, divide the ion between different groups and introduce fractional charges (Evien's method ( Evjen H.M., 1932)).
We will consider the charges on the faces of the cubic crystal lattice (Fig. 2.5) as follows: the charges on the faces belong to two neighboring cells (the charge in each cell is 1/2), the charges on the edges belong to four cells (1/4 in each cell), the charges at the vertices belong to eight cells (1/8 in each cell). Contribution to the α m of the first cube can be written as a sum:
If we take the next largest cube, which includes the one we have considered, then we get , which agrees well with the exact value for a lattice of type . For a type structure, , for a type structure, is obtained.
Let us estimate the binding energy for the crystal , assuming that the lattice parameter and modulus of elasticity IN known. The modulus of elasticity can be determined as follows:
where is the crystal volume. Bulk modulus of elasticity IN is a measure of compression under uniform compression. For a face-centered cubic (fcc) structure of the type, the volume occupied by the molecules is equal to
Then one can write
From (2.53) it is easy to obtain the second derivative
In the state of equilibrium, the first derivative vanishes; therefore, from (2.52–2.54) we define
We use (2.43) and get
From (2.47), (2.56) and (2.55) we find the bulk modulus of elasticity IN:
Expression (2.57) allows one to calculate the exponent in the repulsion potential using the experimental values and . For a crystal , , . Then from (2.57) we have
Note that for most ionic crystals, the exponent n in the potential of repulsive forces varies within 6–10.
Consequently, a large value of the degree determines the short-range nature of the repulsive forces. Using (2.48), we calculate the binding energy (energy per molecule)
EV/molecule. (2.59)
This agrees well with the experimental value of -7.948 eV/molecule. It should be remembered that in the calculations we took into account only the Coulomb forces.
Crystals with covalent and ionic bond types can be considered as limiting cases; between them is a number of crystals that have intermediate types of bonding. Such a partially ionic () and partially covalent () bond can be described using the wave function
in this case, the degree of ionicity can be determined as follows:
Table 2.1 shows some examples for crystals of binary compounds.
Table 2.1. Degree of ionicity in crystals
| Crystal | Degree of ionicity | Crystal | Degree of ionicity | Crystal | Degree of ionicity |
| SiC ZnO ZnS ZnSe ZnTe CdO CDS CdSe CdTe | 0,18 0,62 0,62 0,63 0,61 0,79 0,69 0,70 0,67 | InP InAs InSb GaAs GaSb CuCl CuBr AgCl AgBr | 0,44 0,35 0,32 0,32 0,26 0,75 0,74 0,86 0,85 | AgI MgO MgS MgSe LiF NaCl RbF | 0,77 0,84 0,79 0,77 0,92 0,94 0,96 |